
Overview The GeneList tool allows the user to search for genes using gene IDs, descriptions, experiments, GO ids and different annotations, and then saves the result in a list that can be used by other tools.
Basic Usage
[video width="1600" height="900" mp4="https://help.plantgenie.org/wp-content/uploads/genelist_final.mp4"][/video]
Simply type in gene ids, descriptions, or different annotations. The matching genes will be displayed with selected annotations. The result can be customized by clicking the Select Displayed Annotations button. There are three buttons to Save all to Gene List, Remove selected from Gene List or Empty Gene List. The Share table button allows sharing the current GeneList with other users by way of an auto generated URL. The GeneList tool is the starting point for most PlantGenIE workflow.
Multiple GeneList The GeneList tool is capable of holding several named gene lists for use in other tools. These lists can be Added, Renamed or Deleted. Once clicking on a GeneList name, it will become the active GeneList and will be displayed in all other tools. GeneLists will remain for seven days while shared GeneList will be saved for 30 days.
Data We use both PostgresSQL and MySQL to store annotation data. The GeneLists tool uses both in house annotation data and data from Phytozome and Plaza.
Implementation This tool uses JavaScript, PHP, MySQL, PostgreSQL, JQuery, Datatables and Toastr libraries. SQL views and tmp tables gave additional speed to the tool.

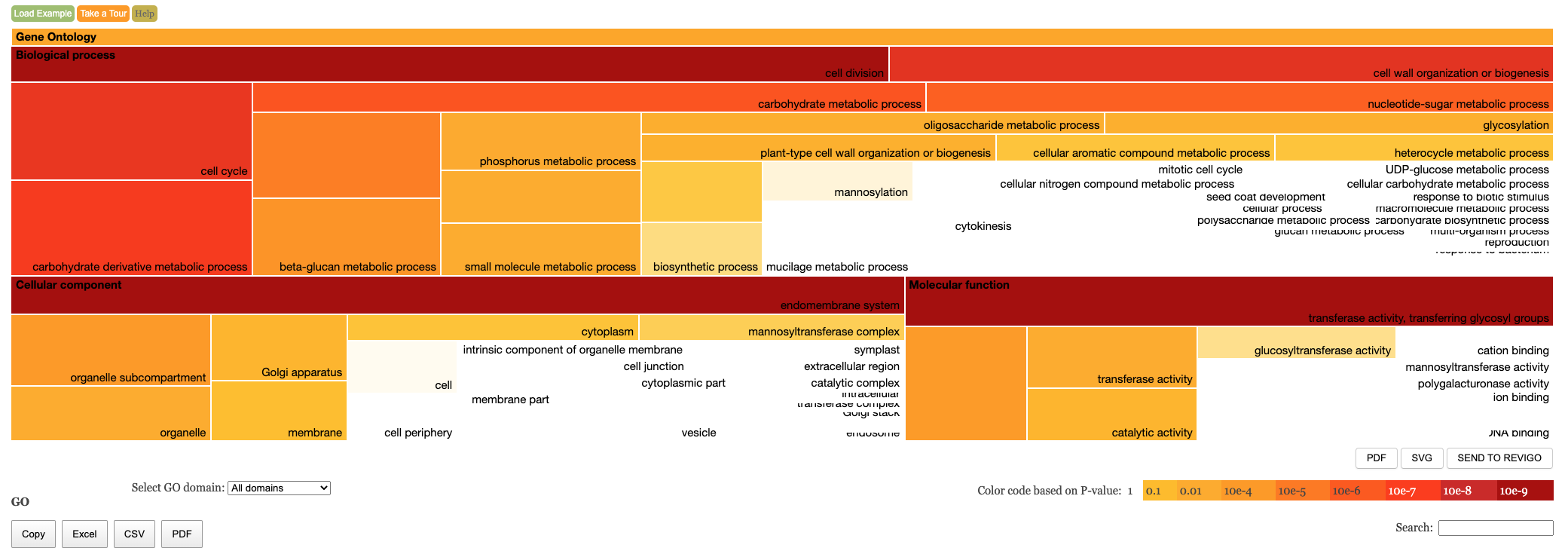
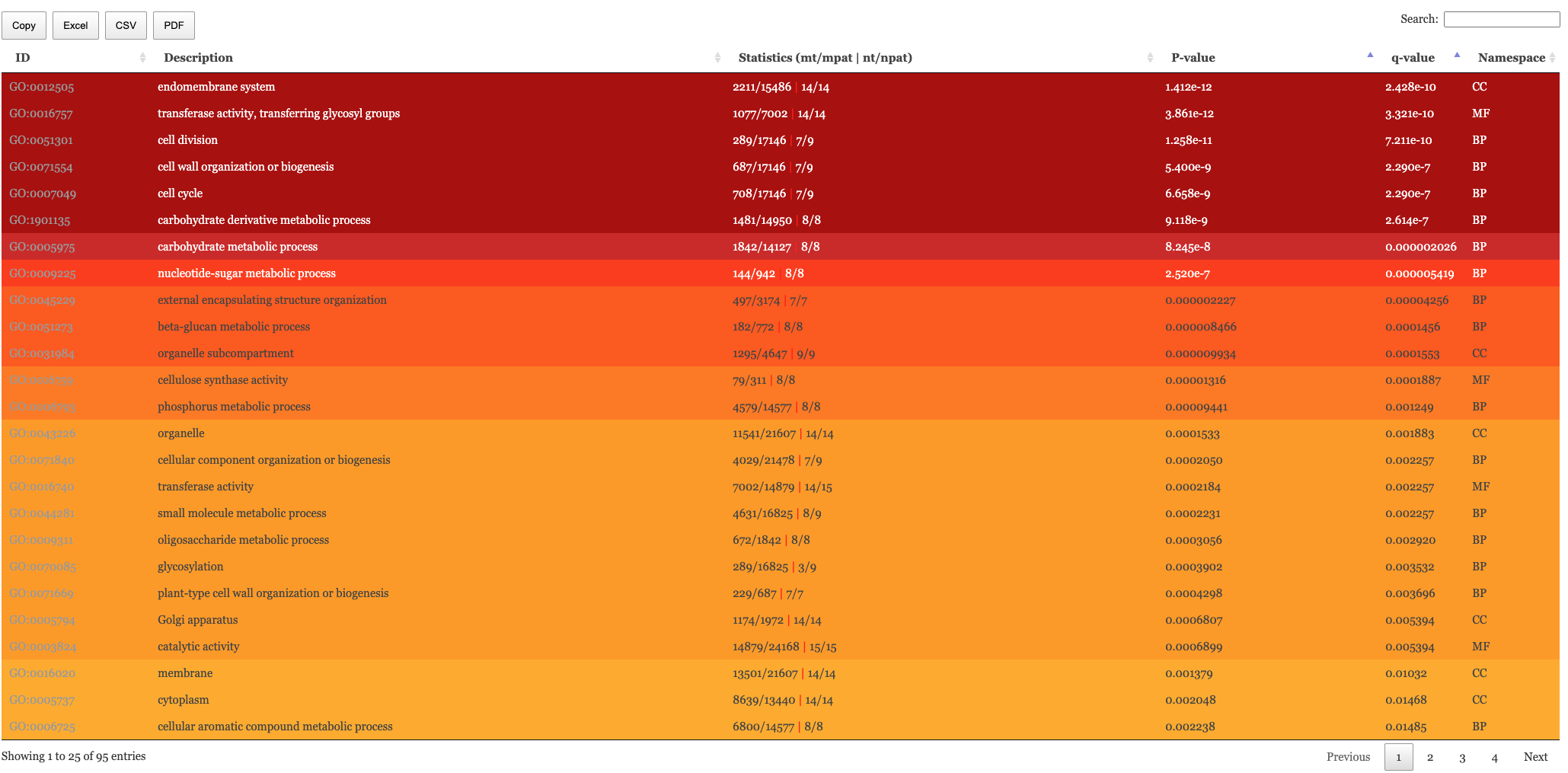 The table of results and tree map figure are colour coded to indicate statistical significance. Red means highly significant and white means less significant. We have the following columns in the Enrichment results.
The table of results and tree map figure are colour coded to indicate statistical significance. Red means highly significant and white means less significant. We have the following columns in the Enrichment results.